
DNA Analysis
 The majority of cells making up the human body are diploid cells carrying identical DNA, with the exception of haploid gametes (egg and sperm) and red blood cells (which have no nucleus). Several types of biological evidence are commonly used in forensic science for the purpose of DNA analysis, including blood, saliva, semen, skin, urine and hair, though some are more useful than others. The use of biological evidence in DNA and genetic analysis varies, with areas of study including blood typing, gender determination based on chromosome analysis (karyotyping), DNA profiling and, more recently, forensic DNA phenotyping. Since the advent of DNA profiling in the 1980s, it has been successfully utilised in criminal cases, disaster victim identification and paternity testing to name a few. However despite their merits, DNA fingerprints are not ideally used as the sole piece of evidence in a case, and in certain countries, such as the United Kingdom, DNA fingerprints must be presented in conjunction with other evidence.
The majority of cells making up the human body are diploid cells carrying identical DNA, with the exception of haploid gametes (egg and sperm) and red blood cells (which have no nucleus). Several types of biological evidence are commonly used in forensic science for the purpose of DNA analysis, including blood, saliva, semen, skin, urine and hair, though some are more useful than others. The use of biological evidence in DNA and genetic analysis varies, with areas of study including blood typing, gender determination based on chromosome analysis (karyotyping), DNA profiling and, more recently, forensic DNA phenotyping. Since the advent of DNA profiling in the 1980s, it has been successfully utilised in criminal cases, disaster victim identification and paternity testing to name a few. However despite their merits, DNA fingerprints are not ideally used as the sole piece of evidence in a case, and in certain countries, such as the United Kingdom, DNA fingerprints must be presented in conjunction with other evidence.
DNA Structure and Function
It is vital to understand the structure and function of DNA and how this relates to DNA analysis in forensic science. DNA, deoxyribonucleic acid, is a molecule arranged into a double-helix, its structure first described by James Watson and Francis Crick in 1953. It is composed of nucleotide trisphosphate molecules, referred to as the ‘building blocks’ of DNA. These molecules consist of a trisphosphate group, a deoxyribose sugar and one of four nitrogenous bases. The four bases involved in a DNA molecule are adenine and guanine (purines) and thymine and cytosine (pyrimidines). These bases bond to the deoxyribose sugar and one of the other bases to form base pairs, with adenine and thymine bonding through two hydrogen bonds, and guanine and cytosine bonding with three hydrogen bonds.

DNA is essentially the molecule that holds all genetic information and ‘instructions’ for an organism. The human genome is composed of over 3 billion base pairs of information organised into 23 chromosomes. Genes are the regions of DNA that encode and regulate protein synthesis, though this involves just 1.5% of the entire genome. A significant amount of the human genome, approximately 75%, consists of extragenic DNA, which contains regions that do not actually contain known gene sequences. About 50% of extragenic DNA is made up of something called repetitive DNA, which is of particular use in forensic DNA analysis. Repetitive DNA is further sub-divided into tandem repeats (including satellite DNA, microsatellites and minisatellites) and interspersed repeats (SINE, LINE, LTR and Transposon). Tandem repeat DNA and the variation between them (polymorphisms) is the focus of many DNA profiling techniques. It is due to the number and location of these polymorphisms that every individual has unique DNA which produces a distinctive band pattern when analysed.
It is through the extensive study of the genome that DNA fingerprinting has been produced as a useful and reliable technique in forensic science.
Sources of DNA Evidence & DNA Extraction
In terms of forensic DNA analysis, there is a variety of possible sources of DNA evidence. The more useful sources include blood, semen, vaginal fluid, nasal secretions and hair with roots. It is theoretically possible to obtain DNA from evidence such as urine, faeces and dead skin cells, though this is often classed as a poor source due to the lack of intact cells and high levels of contaminants preventing successful analysis. Such samples will be collected depending on the type of sample (see crime scenes page for more details of evidence collection and preservation).
Prior to analysis, it will be necessary to extract DNA from the sample. This is generally achieved through the following simplified steps.
- The sample cells are lysed (broken down) in a buffer solution.
- Denatured proteins and fats are pelleted through centrifugation.
- The cleared lysate is then passed through a column, often containing a positively charged medium that binds to the DNA.
- Contaminating proteins, fats and salts are then removed through several washes.
- The DNA is recovered in a buffer solution/water.
- The amount of DNA is often then quantified using spectrophotometric techniques.
- Various methods of extraction have been devised for different types of sample.
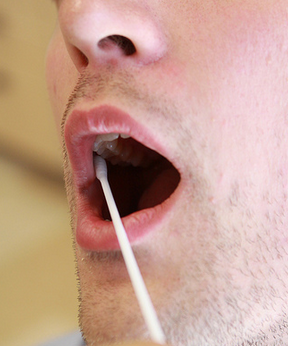
DNA samples for comparison are generally collected from suspects using buccal swabs, in which a sterile swab is scraped along the inside of the cheek to collect epithelial cells to use in producing a DNA fingerprint. Sir Alec Jeffreys is often described as the father of DNA fingerprinting.
Single Nucleotide Polymorphisms
The use of DNA analysis in forensic science is based on a variety of techniques focusing on polymorphisms, which essentially refers to variation in sequences. Different sequences are studied in different techniques, including single nucleotide polymorphisms, minisatellites (variable number tandem repeats), microsatellites (short tandem repeats) and mitochondrial DNA, each different with regards to length and repetition.
Single Nucleotide Polymorphisms (SNPs) are the simplest and most common type of genetic variation, composing around 90% of genetic variation in humans. They occur during meiosis when DNA is replicated, with each SNP representing a difference in a single nucleotide. For example, a SNP may replace cytosine with a thymine in a stretch of DNA, and this will not change the length of the DNA. There are about 10 million SNPs in the human genome, with one found at every 100-300 base pair. These can act as biological markers.
In the analysis of SNPs, which can consist of up the four alleles, the specific base present in the SNP must be established. This is achieved using DNA sequencing techniques. However DNA sequencing is not generally used in forensic science except in the analysis of mitochondrial DNA.
Dideoxy Method
Also known as the Sanger Method, this form of DNA sequencing was developed by Fred Sanger in 1975. A labelled primer is utilised to initiate the synthesis of DNA. Four dideoxy nucleotides are added and randomly arrest synthesis. Fragments are produced which are subsequently separated using electrophoresis or, in more modern automated systems, capillary electrophoresis. Specialised software is then used to convert the band patterns into a DNA sequence. Initially unstable, hazardous radioactive tags were used, soon replaced by fluorescence dyes. However the varying mobility of these different coloured dyes meant that the order of bands did not necessarily accurately represent the order of the nucleotide sequence. The dyes would also sometimes behave unpredictably, making them unreliable and not particularly ideal for use in forensic cases. If conditions were unsuitable for the DNA template or the dideoxy nucleotides, the reaction would be unsuccessful. The computer software itself was problematic, with peaks often overlapping, making it difficult to establish the correct order of nucleotides.
Minisatellites or Variable Number Tandem Repeats (VNTRs)
Variable Number Tandem Repeats (VNTRs) are highly polymorphic sequences in the exon region of DNA repeated at various points along the chromosome, 4-40 times. They are about 20-100 base pairs in length, though their specific lengths are not strictly defined. These tandem repeats are inherited from both parents, therefore no one will have the same VNTRs as either of their parents. The number of repeats at the locus affects its position after electrophoresis and the length of the DNA after the chromosome has been cut with a restriction enzyme. Minisatellites were the first type of polymorphism to be used in a criminal investigation in the case of British murderer and rapist Colin Pitchfork.
However VNTRs are not generally used in forensics, as this technique is often a more expensive process and takes longer due to VNTRs having a greater length than, say, STRs.
Microsatellites or Short Tandem Repeats (STR)
Short Tandem Repeats (STRs) are regions of the genome composed of approximately 1-5 bases and repeated up to 17 times. STR markers will either be simple (identical length repeats), compound (two or more adjacent repeats) or complex (several different length repeats). They are found on 22 autosomal chromosomes as well as both X and Y sex chromosomes, though those on the Y chromosome differ less due to lack of recombination. Only a select number of STR markers are used in forensic DNA profiling (10 in the UK and 13 in the US).They express a high degree of polymorphism, making them of particular use to the forensic scientist. The variability in STRs is caused by the inaccuracy of DNA polymerase in copying the region. As STR regions are non-coding, there is no selective pressure against the high mutation rate, resulting in high variation between different people.
Though there have been thousands of short tandem repeats found in the human genome, only a small number are utilised in forensic DNA analysis. STRs used in forensic science tend to be tetra- and penta-nucleotide repeats, as they are both robust, suffer less environmental degradation, and provide a high degree of error free data. STR loci are ideal for use in forensic science for a number of reasons. They represent discrete alleles that are distinguishable from one another, they show a great power of discrimination, only a small amount of sample is required due to the short length of STRs, PCR amplification is robust and multiple PCR can be used, and there are low levels of artefact formation during amplification. An early use of microsatellites is in the identification of Auschwitz camp doctor Josef Mengele.
Restriction Fragment Length Polymorphisms (RFLPs)
Restriction Fragment Length Polymorphisms (RFLPs) were used in the first technique developed to analyse variable lengths of DNA fragments produced through DNA digestion. It exploits variations in DNA sequences due to the differing locations of restriction enzyme sites. The method uses restriction endonucleases to ‘digest’ the DNA by cutting it at specific sequence patterns. The resulting restriction fragments are then separated using gel electrophoresis and transferred to a membrane using the Southern Blot technique. After the separated DNA fragments are transferred, probe hybridisation is used to detect the fragments.
However DNA analysis with RFLP required relatively large amounts of DNA and degraded samples could not be analysed with accuracy. More effective, faster and cheaper DNA profiling techniques have seen been developed, so RFLP is generally no longer used in forensic science.
Polymerase Chain Reaction (PCR)
The amount of DNA evidence obtained during the investigation of a crime is often very small, thus for successful DNA profiling some form of amplification is ideal. Polymerase Chain Reaction (PCR) is a technique which allows for the exponential amplification of DNA fragments to lengths of approximately 10,000 base pairs. This means that, theoretically, a single copy of a DNA fragment could be amplified to millions of copies in just a few hours. PCR is particularly beneficial in the amplification of minute amounts or degraded samples.
A successful PCR reaction requires a number of vital primary components. Oligonucleotide primers which are complementary to the DNA target and mark the target to be amplified, with two primers being used. The base sequence of one primer binds to one side of the target whilst the other primer binds to the other side of the target, with the DNA between the primers being amplified. Fluorescent tags are often added to the primers to visualise amplified DNA in electrophoresis. DNA polymerase enzyme allows the DNA strand to be copied by adding nucleotides to the 3’ end of the primers. Other components required include a reaction buffer with MgCl to ensure ideal conditions for the functioning of the DNA polymerase enzyme, deoxyribonucleotides to build the DNA molecule, and template DNA. Modern PCR uses thermostable DNA polymerases. Most commonly used is the Taq polymerase, which has largely replaced the previously used E.coli-derived polymerase. This was isolated from Thermus aquaticus, which is an organism capable of surviving in temperatures over 70oC. However Taq polymerase lacks the ability to proof read. VENT polymerase is from Thermococcus litoralis, which can survive in temperatures over 100oC.
The PCR cycle consists of three primary steps: denaturation, annealing and extension. The process is generally conducted in a small, plastic centrifuge tube with the temperature carefully controlled using a thermal cycler.
- Denaturation: The sample is heated to 94-95oC for about 30 seconds. This separates the double-stranded DNA by breaking hydrogen bonds, allowing primers access.
- Annealing: The samples is kept at 50-65oC, depending on the primer sequence, to allow hydrogen bonds to form between the primers and the complementary DNA sequence.
- Extension: Also known as the elongation stage. The sample is heated to 72oC for a duration depending on the length of the DNA strand to be amplified and the speed of the polymerase enzyme (Taq polymerase) which builds up the strand. Deoxynucleotide triphosphates are added to the 3’ end of the primer.
Each PCR cycle can take only 5 minutes. This procedure can then be repeated as necessary until the original sequence has been amplified a sufficient amount of time, with the amount being doubled with each cycle. Following PCR, the products are separated using electrophoresis.
Unfortunately PCR is not suitable in the analysis of longer strands of DNA, and so cannot be used with earlier techniques such as RFLP. It must be taken into consideration that certain compounds can inhibit PCR reactions, often substances associated with the stages of extracting and purifying the DNA. Such substances include proteinase K (which degrades the polymerase enzyme), ionic detergents and gel loading dyes. Similarly, certain substances present in blood can inhibit PCR, such as haemoglobin and heparin.
Various alterations have been made to improve the PCR method. Multiplex Polymerase Chain Reaction involves the amplification of numerous DNA sequences in a single reaction through the use of primers that produce non-overlapping allele sizes, allowing numerous regions of a sample to be tested simultaneously.
PCR Errors
Various factors can contribute to errors and inaccuracies in data produced by the polymerase chain reaction technique. PCR is often carried out using DNA polymerases such as Taq DNA polymerase, which does not have the ability to ‘proof read’, resulting in errors in amplification. The greater the amplification, the more likely it is that such errors will occur. Mispriming is also a potential problem, with products being formed from non-target sites. Excessive primer dimers may be formed, which are by-products of PCR produced when one primer is annealed to another causing primer extension. This may all result in unexpected variability in PCR success across a series of samples or previously successful conditions failing.
Electrophoresis
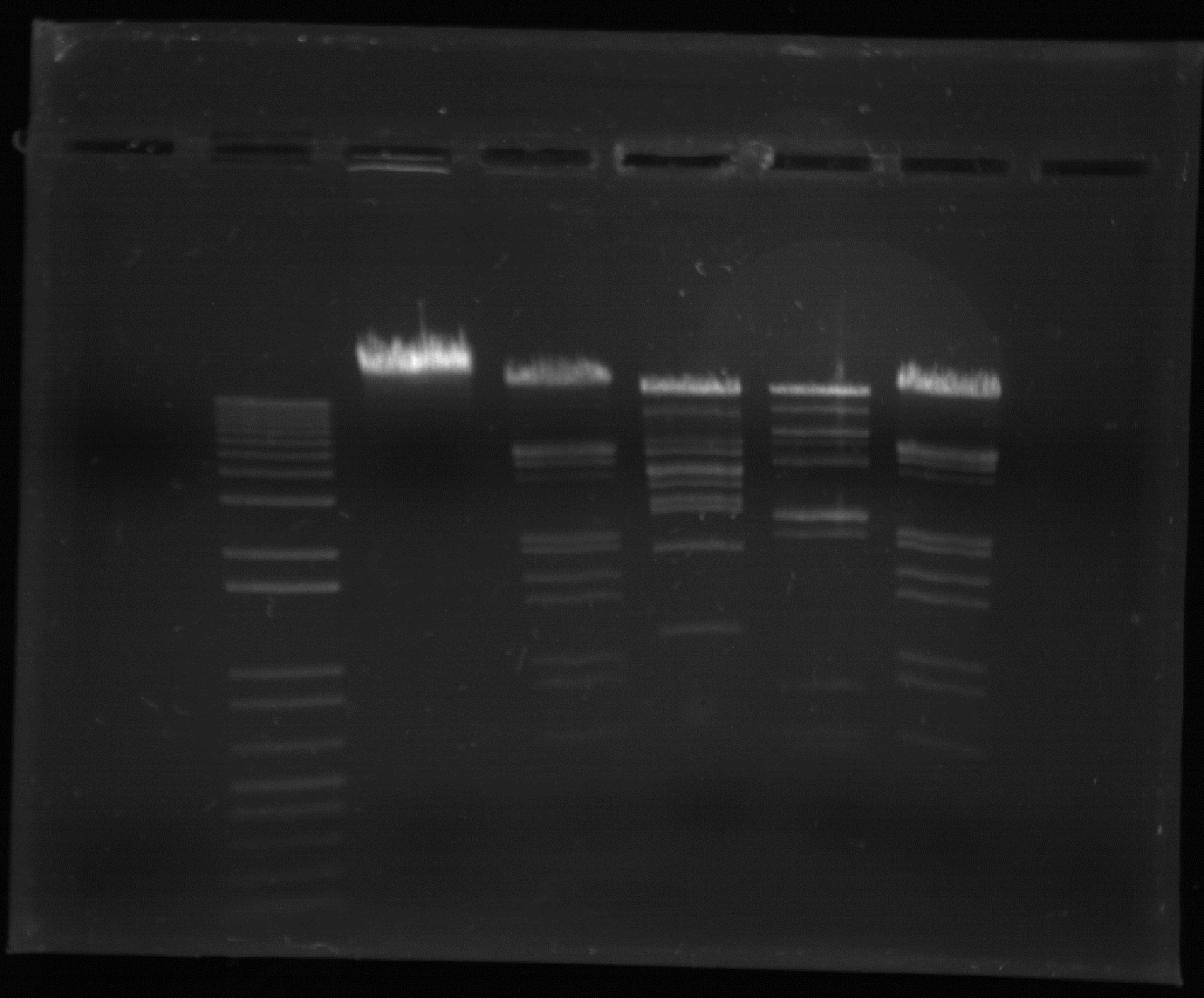
As mentioned, during DNA analysis the individual fragments of DNA can be separated using electrophoresis to produce the distinct ‘DNA fingerprint’. Electrophoresis is essentially a method of separating molecules by their size through the application of an electric field, causing molecules to migrate at a rate and distance dependent on their size. In gel electrophoresis, a porous gel matrix is used, often consisting of agarose gel for simple work or polyacrylamide gel for more specific procedures. The gel is often floating in a buffer solution to ensure the pH level is maintained and the applied electric current is conducted. Samples to be analysed are placed in small wells at the top of the gel using pipettes. A control sample and a standard/marker sample will often be run simultaneously. As the electric current is applied, the negatively charged DNA fragments begin moving through the gel towards the positively charged anode. The gel essentially acts as a type of molecular sieve, allowing smaller molecules to travel faster than larger fragments. Following electrophoresis, it may be necessary to visualise these bands using radioactive or fluorescent probes or dyes. Electrophoresis not only separates DNA but also allows for the fragments to be measured, often expressed in base pairs. Measuring the length of these fragments can ultimately allow the number of repeats to be determined and thus the genotype at that locus.
Earlier techniques used flat-bed gel electrophoresis, though the faster and automated capillary electrophoresis is now used more often. In this technique, abbreviated to CE, the gel is held in a fine capillary tube through which the fluorescently-labelled DNA passes through much as in gel electrophoresis, often with an added DNA size standard. Positioned along the capillary is a laser beam which causes the DNA to fluoresce as it passes. This can then be detected and fed directly to a computer system in the form of an electropherogram. Unfortunately capillary electrophoresis is not able to separate more than one sample at a time, though a genetic analyser can be used to separate a series of samples one after the other.
Blotting Techniques
Following gel electrophoresis, probes are generally used to detect specific molecules. However because probes cannot be directly applied to the gel, blotting methods were initially utilised. There are a number of common blotting techniques: Southern blot, Western Blot, Northern Blot, Eastern Blot and Southwestern Blot.
- Southern Blot – Used in DNA analysis. DNA is extracted, separated with electrophoresis and transferred to the membrane. Labelled probes are added, which hybridise to specific sequences and identify them.
- Western Blot – Used to detect proteins. SDS-PAGE used to separate proteins, which are transferred to membrane. Specific antibodies are then added, followed by a substrate to visualise bands.
- Northern Blot – Used to study gene expression. Similar to the Western blot, except RNA is being analysed.
- Eastern Blot – Used to study proteins. Considered to be an extension of the Western blot.
- Southwestern Blot – Combines features of the Southern and Western blot. Used for the rapid characterisation of DNA binding proteins and their sites.
Low Copy Number (LCN) DNA Analysis
Low Copy Number DNA Analysis, referred to as LCN, is a technique developed by the UK’s Forensic Science Service in an attempt to increase the sensitivity of DNA profiling methods. Samples containing small amounts for badly degraded DNA often leads to problems such as poor quality fingerprints or even completely negative results. This technique reduced these issues.
Developed in 1999,LCN is essentially an extension of the Second Generation Multiplex Plus (SGM+) technique, and is generally used when the previous technique has failed or has produced weak results. Improved sensitivity is achieved through an increased number of PCR cycles, with standard techniques generally using 28 cycles but LCN using 34. This could ultimately allow for DNA profiles to be successfully obtained from minute amounts of sample and even from single cells (see below).
However the increased sensitivity of this DNA profiling technique brings about amplified concerns over issues of ease of contamination and amplification of these contaminants, mixed profiles being produced and wrongful accusations. With techniques such as LCN, it is now more important than ever that investigators wear suitable protective clothing and follow strict anti-contamination procedures, and controls are used in analyses.
In 2007, LCN came under great scrutiny through the case of R vs Hoey, which led to the use of this technique being temporarily banned until a thorough review could be conducted. Sean Hoey was tried for involvement in the Omagh bombing in 1998, charged with numerous counts of murder in addition to terrorism and explosives charges. The DNA evidence used against him was based on the relatively new LCN technique which, at the time, was only being used in British courts. However due to the lack of data and known error rates regarding the technique, serious concerns were raised. Hoey was subsequently found innocent.
Single Cell DNA Fingerprinting
Closely linked to LCN analysis is single cell DNA profiling. Dr Ian Findlay and his colleagues at the Australian Genome Research Facility first reported the successful development of a DNA fingerprint from a single cell in 1997. 226 buccal cells from four individuals were isolated using micromanipulation, amplified using the UK Forensic Science Service’s Second Generation Multiplex (SGM) assay to increase sensitivity. Six STR markers and amelogenin for sex-typing were amplified. The results from single-cell analysis were compared with known DNA profiles. Whereas a full and correct profile was only obtained in 50% of the single-cell tests, 91% of tests exhibited some form of result.
Previous DNA fingerprinting methods often required hundreds of cells in order to obtain a profile. The single cell is obtained by swabbing the material and identifying the cell to be analysed using microscopy prior to analysis. This technique is particularly fast, taking a matter of hours, and has a specificity of about 1 in ten billion.
Single-cell DNA profiling is particularly useful in rape cases, as DNA in sperm cells is highly conserved due to it being so compacted in the protein head. There is also potential for the technique in use in documents. Human DNA can be placed in documents such as Government bonds, wills and security documents, to track their flow. However the main issue with this particular use is that close relatives may handle the documents, particularly when dealing with documents such as wills, and so the technique may not be appropriate. Single-cell DNA analysis is also ideal in IVF procedures, in which single cells can be analysed for genetic defects.
However there are some major concerns with this method. As an increased number of PCR cycles are required to amplify the DNA, this brings about problems of allele drop-in, where additional alleles are added to the sample. Allele drop-out is a similar problem with increasing amplification samples. When starting with a single cell or small amount of sample, any contaminants already present in the sample will be amplified. Similarly, any PCR inhibitors present will be more concentrated, causing amplification problems. One of the primary concerns is the ease of contamination by cells from other individuals. This could result in samples being contaminated and rendered useless or, worse still in the case of forensics, innocent individuals being wrongly accused. More work is required to validate the technique, particularly to use it in forensic science.
The work in single-cell DNA analysis led to the Forensic Science Service in the UK developing low-copy number DNA analysis.
Mitochondrial DNA (mtDNA)
Mitochondrial DNA is a circular molecule of DNA 16,569 base pairs in size, first referred to as the Anderson sequence, obtained from the mitochondrion organelle found within cells. This organelle is involved in the production of cell energy. There can be anywhere between 100 and 1000 mitochondria within a cell, each one containing numerous copies of the mitochondrial genome. This sequence is entirely functional and highly conserved, so there is very little variation between individuals. However there is a 1000 base pair long non-coding D-loop, known as the control region, which contains two hypervariable regions referred to as HV1 and HV2. The variations within these regions are generally single nucleotide polymorphisms (SNPs). SNPs do not alter the length of the mtDNA, and it is these regions that are focused on in the forensic analysis of mitochondrial DNA. Mitochondrial DNA is often subject to a relatively high rate of mutation due to its lack of DNA reparation, causing variation between individuals. The variation within this small portion is itself not especially significant, with HV1 and HV2 differing by 1-3% between non-related individuals.
In the analysis of mitochondrial DNA, the DNA is extracted and the HV1 and HV2 regions amplified using PCR. The base pair sequence of these regions is then established through DNA sequencing (see DNA sequencing section). This is then compared with the Cambridge Reference Sequence and differences noted. Other samples can then also be analysed an comparisons made to establish potential similarities. Mitochondrial DNA is generally used when other methods such as STR analysis have failed. This is often in the case of badly degraded bodies, in cases of disaster or accidents where an individual is too badly damaged to identify, and sometimes in taxonomy to determine species using the cytochrome b gene.
The most significant advantage of the use of mitochondrial DNA is the possibility of analysing even highly degraded samples. If a specimen is severely decomposed to the point that it is not possible to successfully extract a DNA profile using nuclear DNA, it may be possible through mitochondrial DNA. Additionally, only a very small sample size is required.
However the use of mtDNA does have its disadvantages. As mitochondrial DNA is only maternally inherited, this cannot form a full DNA fingerprint of the individual, thus this technique is only beneficial if the DNA profiles of maternal relatives are available, such as the individuals mother or biological siblings. Because of this, mtDNA is significantly less discriminatory than, for example, Short Tandem Repeats. Detecting sequence differences is also relatively time consuming and expensive.
Forensic DNA Phenotyping
Attempts have been made to utilise DNA analysis in the identification of phenotypic characteristics such as skin colour, hair colour and eye colour in a study known as forensic DNA phenotyping (FDP) or phenotypic profiling. This is generally achieved using single nucleotide polymorphisms (SNPs) rather than STRs. SNPs have a lower mutation rate and so are more likely to become fixed in a population, thus they are often found to be population-specific. It may be possible to estimate ethnic origin based on the presence of rare SNPs or STRs linked to particular population groups, though this theory becomes problematic with individuals of mixed ancestry.
Most work on phenotype SNPs has focused on pigmentation, and SNPs in a number of pigmentation genes have been associated with various human hair, skin and eye colour phenotypes.
Advances are already being made in this area of study. The Forensic Science Service has developed an SNP typing assay involving mutations in the human melanocortin 1 receptor gene (MC1R), which is associated with red hair. Specific alleles along this gene are associated with red hair, thus an individual inheriting this allele from each parent results in a high likelihood of that individual having red hair. Establishing that an individual is likely to have red hair is limited in forensic science, as red hair is not particularly common, though it is more common to certain populations.
Some research has been carried out into the genetics of eye colour, namely relating to the OCA2 gene on chromosome 15, which is also involved in the pigmentation of both skin and hair. IrisPlex is a recent test developed which allows for the accurate prediction of blue and brown eye colour.
Studies have been conducted examining the frequency of specific short tandem repeat alleles in groups of varying geographic ancestry. This research has resulted in the possibility of likely ancestry being established due to certain alleles being more likely in specific groups. However this particular use of DNA analysis is not infallible and can only be used as an estimation. The Forensic Science Service has developed an ethnic inference test, which provides the likely origin of a DNA sample from a number of groups (white European, Afro-Caribbean, Middle Eastern, South-East Asian and Indian Sub-continental).
Some countries and states are implementing specific legislation relating to the use of phenotypic DNA analysis. Many jurisdiction presently only allow the analysis of non-coding DNA, though the Netherlands currently allows forensic phenotyping under certain regulations. The phenotypic use of SNPs have also been used in non-forensic analysis, such as in determining the ethnicity, hair and skin colour of ancient remains.
However there are a number of problems and concerns with this approach. It is unlikely that a few chosen SNPs will provide a foolproof ‘picture’ of the sample’s source due to the complexity of multigenic traits as well as external factors such as environment and aging. Even if the technique was perfected for use in forensic science, phenotypes such as hair or eye colour can easily be masked through dyes and coloured contact lenses, limiting its forensic use. There are also privacy concerns relating to the possible traits determined, though it has conversely been argued that visible traits such as hair and eye colour cannot be considered private. Though should further advances be made to the extent that genes were located for traits such as aggression and predispositions to certain diseases, more serious concerns would be raised over the sensitivity of such information.
Researchers anticipate that further advances could allow for additional details to be ascertained, such as the likelihood that the individual smokes, along with the possibility for genes for the likes of handedness, aggression and homosexuality.
Y Chromosome Analysis
Much of modern DNA profiling is based on the analysis of short tandem repeats found on autosomes. However one particular branch of DNA analysis focuses on the amelogenin marker, the only marker on the sex chromosome, useful in the analysis of the Y chromosome. The Y chromosome, generally found only in males, is a small chromosome which, unlike other genes, is only altered through the infrequent occurrence of mutation. However similar to mitochondrial DNA (which is maternally inherited), the combination of alleles in this instance is theoretically identical between father and son, assuming mutation does not occur. Furthermore, Y chromosome analysis discrimination is comparatively low. Y chromosome analysis is particularly useful in cases of sexual assault and rape in which mixed DNA profiles may be encountered. Numerous systems have been developed to analyse some of the STRs present on this chromosome, such as Applied Biosystems’ Yfiler.
DNA Databases
Numerous countries have produced computerised databases containing DNA profiles to aid in the comparison of DNA fingerprints and the identification of suspects and victims. The first Government DNA database was established in the United Kingdom in April 1995, known as the National DNA Database (NDNAD). As of 2011, there were over 5.5 million profiles of individuals in the system. Similarly, the FBI in the US formed their own DNA database, the Combined DNA Index System (CODIS), in 1994, though it was not implemented in all states until 1998. Staff members involved in the handling and analysis of evidence will often also submit their DNA profiles to the database in the case of accidental contamination. There is the possibility for DNA databases to be shared between countries, however some countries focus on different loci in DNA fingerprinting.
DNA databases are not only used to make direct matches between the DNA fingerprints of one person, but to also conduct familial searching. This involves the search for genetic near-matches between a victim/suspect and a member of their family whose DNA profile is stored. This technique is based on the principle that related individuals are likely to express similarities in their DNA profiles. The first prominent use of familial searching was in the case of British serial killer Joseph Kappen, who was seized after his son’s DNA profile was obtained and then linked to him.
The production of DNA databases has allowed for the faster apprehension of suspects through comparing new crime scene samples to those already stored in the database, providing links between criminals and other crimes. It has also been widely used in cold cases, in some instances proving the guilt of an individual decades after they committed the crime. Conversely, wrongly imprisoned individuals have been exonerated through the advent of new DNA analysis techniques and databases. There is also the potential benefit of identifying bodies that have been too badly damaged or decomposed to identify, provided the individual’s DNA profile is stored.
However despite the advantages of such databanks, there has been significant controversy relating to the subject. Standard practice in many locations is to take a DNA sample of an individual when arrested, but this raises concerns over whether their DNA should be retained if they are then found innocent. There are also worries over innocent people being identified as matches or partial matches to DNA found at a crime scene even if they were not involved in the crime but had innocently attended the scene at some other point in time. This particular problem is becoming increasingly concerning as DNA fingerprinting techniques become more sensitive. There are also privacy concerns over the availability of sensitive genetic information, such as susceptibility to certain diseases and familial relationships. A more administrative disadvantage of such databases relates to the need for a facility that is both large enough to store such data but also has adequate security, a combination that can prove extremely expensive.
DNA Profile Interpretation
The primary purpose of forensic DNA profiling is to obtain a DNA ‘fingerprint’ from a biological sample and compare this to profiles obtained from DNA from a crime scene, an individual or profiles stored on a database. The process of modern DNA profiling includes statistically determining the chance that two people will share the same profile by establishing how common a genotype or collection of genotypes is within a population. The more loci studied and the greater heterozygosity of these loci, the smaller the chance two people will share a profile. However such figures can only ever be estimations and do not take certain factors into consideration, such as biological relatives. DNA evidence tends to be presented in terms of a random match probability, rather than definitively stating whether two profiles match or not.
Jeffreys, A J. Thein, S L. Wilson, V. Individual-Specific Fingerprints of Human DNA. Nature. 1985, 316)6023), 76-79.
Watson, J D. Crick, F H. A Structure for DNA. Nature. 1953, 171, 737-738.
Findlay, I. Taylor, A. Quirke, P. et al. DNA Fingerprinting from Single Cells. Nature. 1997, 389(6651), 555-556.
Jackson, A. R. W, Jackson, J. M., 2011. Forensic Science. Essex: Pearson Education Limited.
White, P. C., 2004. Crime Scene to Court: The Essentials of Forensic Science. Cambridge: The Royal Society of Chemistry.